
Staying abreast of regulatory changes can be a game changer for your business

Are you a football fan? (To be honest, I’m not, really.)
But regulatory football? Well, that’s something else entirely.
Why do you care about regulatory change? Keeping pace with change generates cash flow. Your product gets to market faster and stays on the market longer. When you’re doing those things, you’re making money.
For FY 2018, the U.S. Food and Drug Administration’s (FDA) Center for Devices and Radiological Health (CDRH) plans on issuing 20 new guidance documents. Most of these are pretty wide-ranging and are going to apply to most every medical device company. Unfortunately, the FDA’s agenda is the shortlist compared to the regulatory changes in Europe and the rest of the world.
So many changes are happening that some large consulting firms decided to throw in the towel for the medical device sector of their business, citing “the perfect storm of dramatic changes being driven by new FDA regulations, and European Commission directives for CE Marking. Simply put, the simultaneous changes put dramatic pressures on medical device manufacturers, all at once, and … feels it no longer has the appropriate in-house expertise to support all these requirements.”[1]
If you are reading this article, you are likely fairly entrenched in the medical device space; throwing in the towel and changing market sectors are not options for your business model. Perhaps your company is taking the ostrich or wait-and-see approach to compliance before implementing the new requirements. Perhaps your company has been under the proverbial rock and, prior to reading this, was blissfully unaware the regulatory world as you know it is coming to an end.
If you have taken this approach to compliance, you may have unwittingly thrown in your towel as well. It may already be too late in the game for you to retain certifications needed to meet new requirements.
So, What Can You Do to Understand and Keep Up With These Changes?
This article focuses on how these changes impact your business, why you should care, and some tools that you can put in place to be better prepared in your business strategy.
How did we get all of these new regulatory requirements? For a moment, let’s pretend that navigating the changing regulatory environment is like playing a football game. What does that field look like for the medical device sector?
In any football game, you’ve got the key players on the sidelines that are watching that field of play. For medical devices, the people who are forming those boundaries of the field of play are your regulators — the FDA, the European Commission, and a number of other countries that have formal medical device regulations. And then there’s always a set of customer requirements that may or may not align with those regulatory expectations.
Now, when medical device companies were first regulated, these boundaries were very loosely, if it all, tied together. They were high level and very generic, as in have a quality system, validate your processes, look at your complaints. The guidelines weren’t overly prescriptive about how those activities had to be done.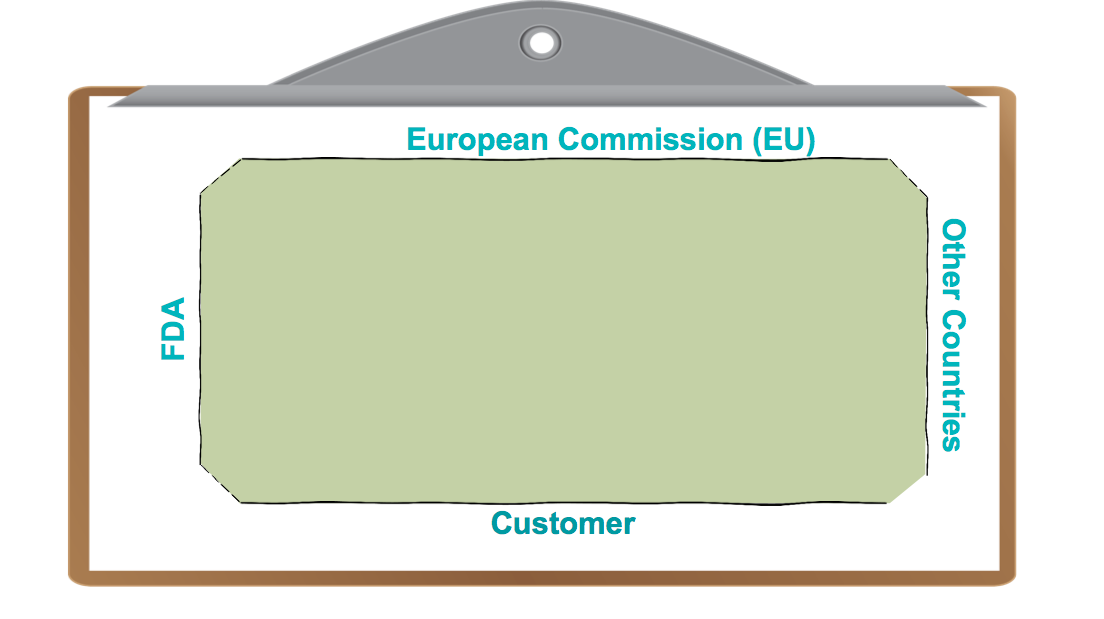
Figure 1 – For medical devices, international regulators and customers define boundaries for the playing field. In early years of regulation, these boundaries were very loosely, if it all, tied together.
As medical device companies started playing this game, some of them chose not to play inside that field altogether. Some of them bent the rules so far that the playing field didn’t look the same anymore.
In those scenarios, companies caused incidences for patient harm. When patient harm happens, the European Union, the FDA and those other regulatory bodies get up and they say, “You know what, we gave you enough rope and all you did was hang yourself. So now we’re going to get real prescriptive with what compliance looks like.”
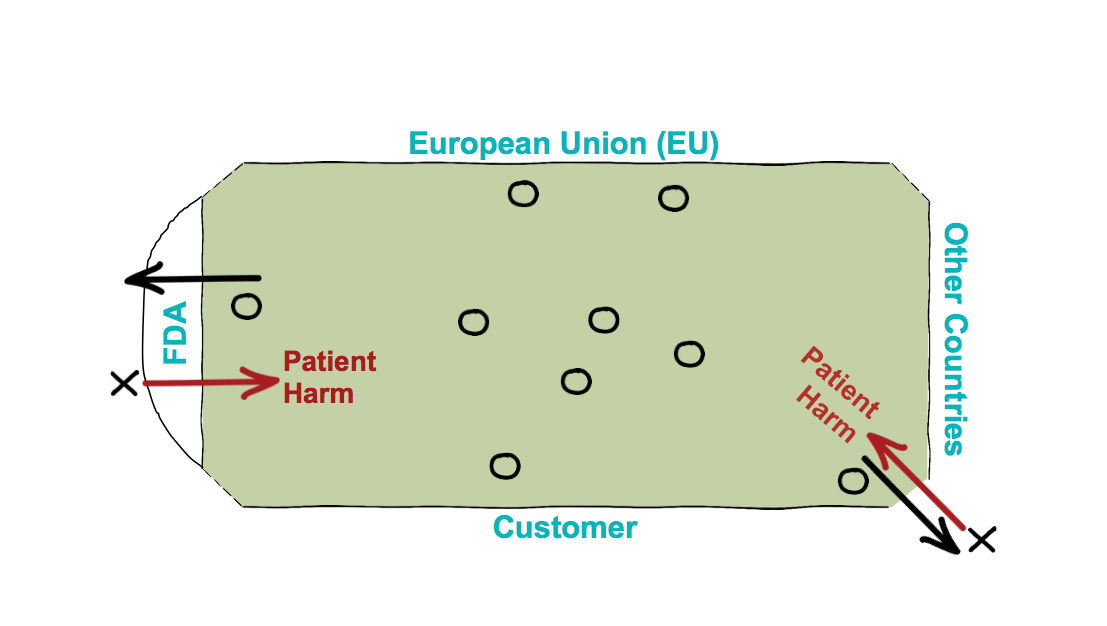
Figure 2 – Many medical device companies chose to bend the rules of the game. Others chose to play outside the bounds all together. In such scenarios, companies caused incidences for patient harm.
As a result, the FDA starts issuing those 20-guidance documents a year. They start the initiatives for the unique device identifier so that they can better tie those patient harm incidences back to specific medical devices.
The EU redrafts the medical device directive to the medical device regulations. They also have their own set of guidance documents that they expect you to implicitly fulfill.
And then lastly, there’s a set of ISO standards that govern not only your quality management systems, but your product performance criteria. The FDA is rapidly moving to recognize these standards.
So, what does it mean for your playing field? Well, obviously, it’s not the same shape or size it used to be. These external demands exert pressures that make the boundaries difficult to define. Playing inside the bounds makes navigating what’s in bounds and out of bounds confusing, which means it’s easier to cause patient harm, and it’s also easier to cause a regulatory incident.
But Why Do You Care?
The playing field is where your cash is. If you get to market faster and stay on the market longer, you’re making money. Being current with regulatory change helps you know where those boundaries on the field of play are so that you stay in that profitable zone.
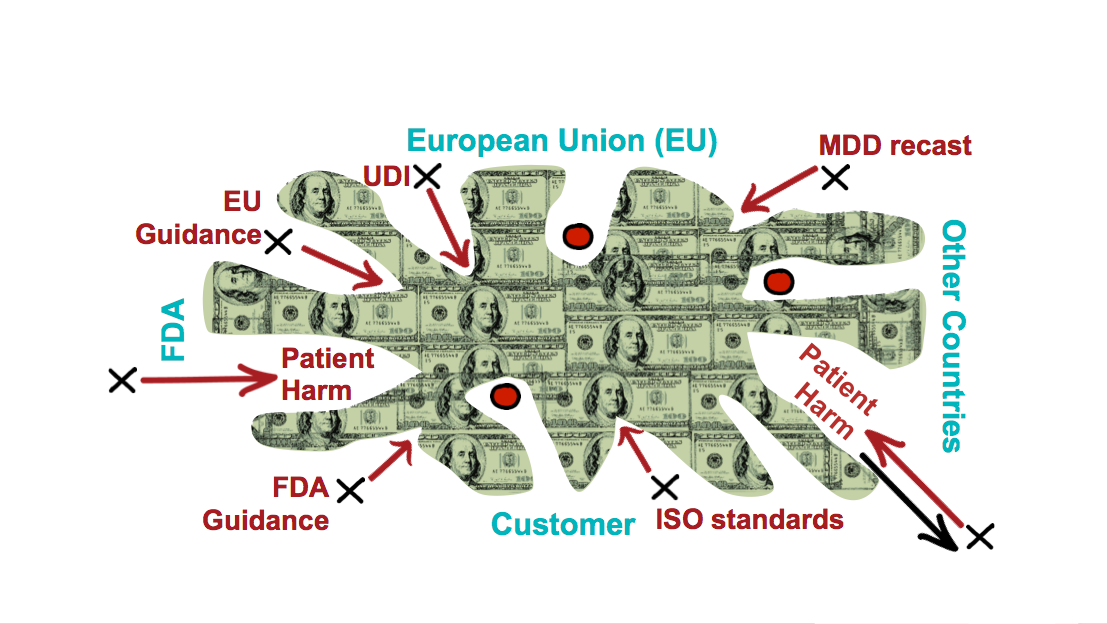
Figure 3 – Pressures from new requirements change the shape of the playing field but keeping the play in bounds is where your cash is. If you get to market faster and stay on the market longer, you are making money. Being current with regulatory change helps you know how to stay in that profitable zone.
Calling the Plays
How do you stay current with the regulatory playbook? First off, check in. Be aware. Know what politics are changing around your technology. Know what guidance documents or regulatory changes are going to impact your business strategy and do a gap analysis so that you can make a plan.
If you don’t fully understand how a changing guidance affects your product, ask the agency directly in charge to explain how it will affect you. Is a physical product alteration required or is the regulatory body’s perception of a technology impacting your product? Your cheese has moved. Don’t fight it.
Your regulatory employees are not embellishing when they inform you of the coming regulatory tsunamis. They have other things to do for hobbies besides creating new regulatory requirements. Be proactive. Figure out how you can work new requirements into your business strategy as a competitive advantage.
And then, finally, know your basics because all of these policy and regulatory changes could directly impact the way you currently understand your product and its requirements. Understand all of the information about your product, how it is regulated, test requirements, and specifications readily available. The changing requirements may even reclassify your product. If you do not already understand the lay of the land, you will not be able to navigate the changing terrain.
Do Those Basics and You’ll …
… Increase the safety and efficacy of your device, which is going to help you better meet your patient and market needs. Your products will get to market faster with better longevity. You’ll build brand credibility with your customers and regulators. And when you get the basics right, you’re safely navigating the boundaries of the field of play, staying squarely where you’re making cash and being profitable.
References
- https://www.oxebridge.com/emma/oxebridge-stops-offering-iso-13485-implementation-citing-perfect-storm-changes/
About the Author:
 Michelle Lott has been the Principal and Founder of Lean RAQA since 2010. By 2018, Lean RAQA supported more than 100 companies with services including regulatory strategy, regulatory submissions, quality systems compliance, due diligence, and a considerable range of technical support services. Lean RAQA specializes in supporting startups’ first quality system and submission as well as remediation services for large companies. Michelle currently serves a four-year term on the FDA Device Good Manufacturing Practices Advisory Committee (DGMP); reviewing feasibility and reasonableness, recommending proposed good manufacturing practice regulations in manufacturing, packing, storing, and installing devices. Michelle achieved a Regulatory Affairs Professionals Society Executive Leadership Certification from Northwestern Kellogg School of Management and a BS in Chemistry from Troy State University. Interesting fact: Michelle formerly served as an expert witness in forensic toxicology at the Mississippi Crime Laboratory.
Michelle Lott has been the Principal and Founder of Lean RAQA since 2010. By 2018, Lean RAQA supported more than 100 companies with services including regulatory strategy, regulatory submissions, quality systems compliance, due diligence, and a considerable range of technical support services. Lean RAQA specializes in supporting startups’ first quality system and submission as well as remediation services for large companies. Michelle currently serves a four-year term on the FDA Device Good Manufacturing Practices Advisory Committee (DGMP); reviewing feasibility and reasonableness, recommending proposed good manufacturing practice regulations in manufacturing, packing, storing, and installing devices. Michelle achieved a Regulatory Affairs Professionals Society Executive Leadership Certification from Northwestern Kellogg School of Management and a BS in Chemistry from Troy State University. Interesting fact: Michelle formerly served as an expert witness in forensic toxicology at the Mississippi Crime Laboratory.